Crystal Structure Producer (CrySP)
A Python package developed for automated crystal structure creation of layered metal-organic frameworks
Crystal Structure Producer (CrySP)
An in-house Python package, coined Crystal Structure Producer (CrySP), is developed to create periodic structures of EC-MOFs. The CrySP algorithm starts by rotating the organic linker to the desired position, then functional groups are placed around the organic linker according to the connectivity of the linker. Next, metal nodes are added according to the bond length between metals and functional groups which is specified in advance. After necessary transformations, the structure is rotated to fit into the specified unit cell. Some specific details can be different between different classes of organic linkers that are defined in EC-MOF/Phase-I database, as shown in the figure below. At the same time, CrySP calculates cell parameters for each class of materials by taking the positions of metal nodes as reference points.
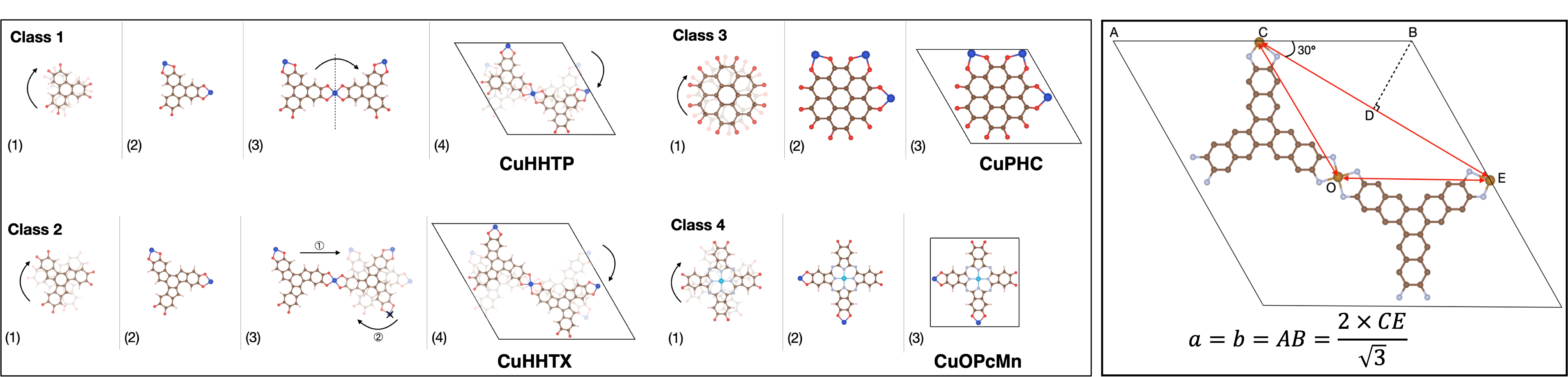
High-throughput Screening (HTS) of EC-MOF/Phase-I database
An HTS workflow is constructed to implement density functional theory (DFT) calculations with periodic boundary conditions, which are all carried out using Vienna ab initio simulation package (VASP) version 5.4.4., [1] to optimize the structures gathered in EC-MOF/Phase I database and to calculate some of their most significantly relevant properties. We have developed a user-friendly platform here where the users can visualize and download the cif/XYZ/POSCAR files of the optimized EC-MOFs. Upon the choice of desired metal nodes and organic linkers, the HTS calculated properties of the optimized EC-MOF is displayed at the bottom of the page.
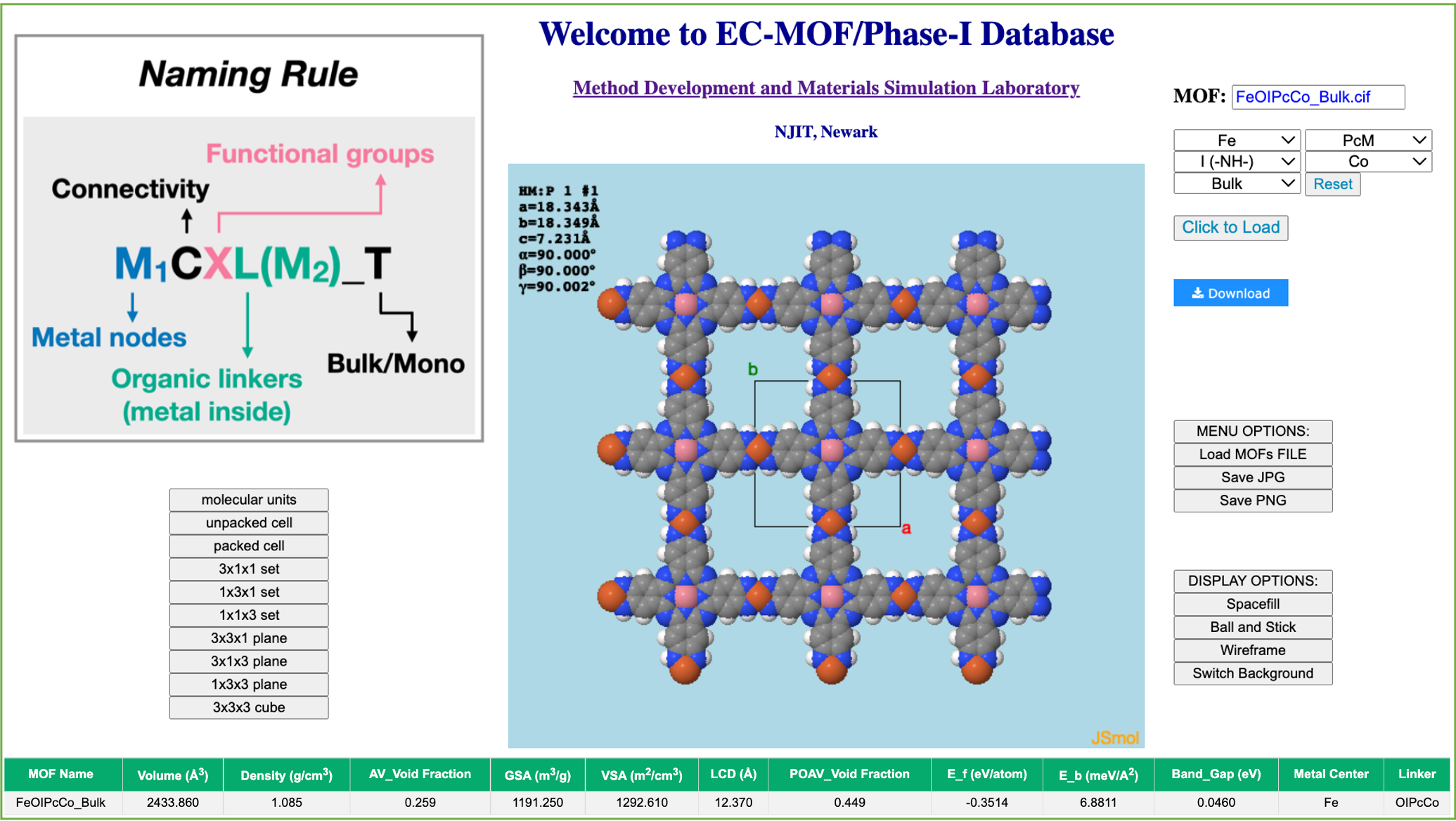
References:
[1] (a) Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561; (b) Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–
amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269; (c) Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50; (d) Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186.